Jornalismo Independente










Proposta do governo prevê reajuste do limite de faturamento do MEI para corrigir a inflação acumulada desde 2018 e ampliar o alcance do regime.



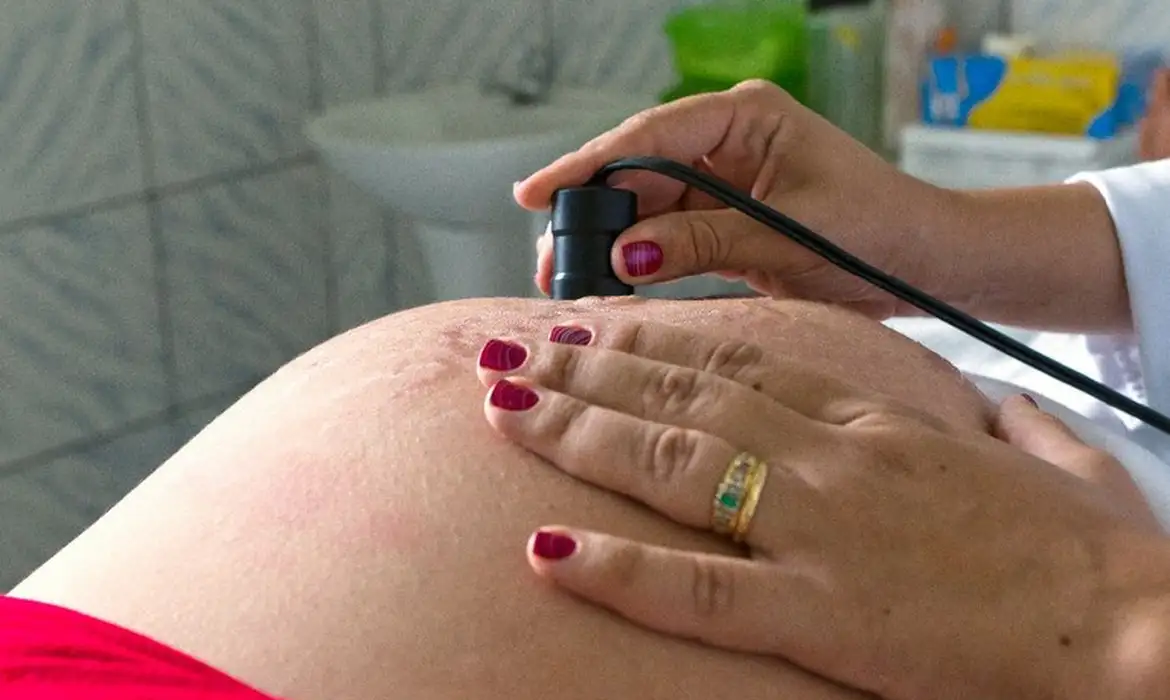
Número de brasileiros que contribuem para a Previdência chega a 68,4 milhões no trimestre encerrado em maio, segundo o IBGE
Conversas completas e leituras para quem valoriza contexto, tempo e profundidade.















📩 Descubra o mundo pelos olhos de Mara Luquet! A fundadora do MyNews compartilha insights exclusivos sobre longevidade, política, economia e finanças pessoais diretamente de suas vivências no Brasil, na Costa Rica, em Lisboa e em suas viagens pelo país e pelo mundo. Na newsletter ‘Recado da Mara’, você encontra histórias que não chegam aos jornais, análises afiadas e reflexões de quem vive o jornalismo além da redação. Assine e acompanhe!
 Mercado Financeiro
Mercado Financeiro
 Mercado Financeiro
Mercado Financeiro
Mercado Financeiro
Mercado Financeiro
 Mercado financeiro
Mercado financeiro
 Benefícios sociais
Benefícios sociais
 Conflito no Oriente Médio
Conflito no Oriente Médio
 cinema brasileiro
cinema brasileiro
 Sustentabilidade
Sustentabilidade
 oriente médio
oriente médio
Presidente dos EUA pressiona Teerã por concessões sobre urânio, Estreito de Ormuz e apoio a grupos armados no Oriente Médio
Continuar lendo
Estudo identifica mais de 300 incidentes com danos ambientais e alerta para impactos globais do conflito
Continuar lendo crise humanitária
crise humanitária
 Sustentabilidade
Sustentabilidade
 CRIME AMBIENTAL
CRIME AMBIENTAL